
Company advances global neuro-intervention footprint with new CE-marked devices for vasospasm and stroke, and expanded U.S. indications for aspiration catheters. PLANO, Texas, Feb. 10, 2026 /PRNewswire/ — Vesalio, a global leader in vascular intervention, today announced CE Mark…
Regulatory
Peijia Medical Submits EU MDR CE Mark Registration Application for GeminiOne® TEER System
HONG KONG, Feb. 9, 2026 /PRNewswire/ — Peijia Medical Limited (HKEX: 9996) today announced that it has formally submitted the European Union Medical Device Regulation (EU MDR) CE Mark registration application for its internally developed GeminiOne Transcatheter Edge-to-Edge Repair (TEER)…
Abbott receives CE Mark for the TactiFlex™ Duo Ablation Catheter to treat patients with abnormal heart rhythms
Abbott’s TactiFlex™ Duo Ablation Catheter, Sensor Enabled™, is designed with dual-energy to treat atrial fibrillation patients with the most challenging cases It can deliver both radiofrequency energy and pulsed field ablation (PFA) energy during procedures to target and treat an…

AliveCor Announces FDA Clearance of New Cardiac Determinations for Kardia 12L ECG System, Bringing Total to 39
KAI 12L, the AI powering Kardia 12L, adds five new detections to further expand diagnostic capabilities for the world’s first handheld 12-lead ECG with a unique single-cable design. KAI 12L, the AI powering Kardia 12L, adds five new detections to further expand diagnostic capabilities for the world’s first handheld 12-lead ECG with a unique single-cable design.

RAMPART Launches SECURE Clinical Program to Advance Clinical Evidence for Next Generation Radiation Protection
BIRMINGHAM, Ala. — January 8, 2025 — Rampart, a Birmingham, Alabama–based medical device company redefining interventional radiation safety, today announced the launch of the SECURE Clinical Program, Studies Evaluating Clinical Utility of Radiation mitigation Equipment, a first-of-its-kind, multi-specialty clinical initiative designed to further advance the scientific evidence for next-generation radiation […]
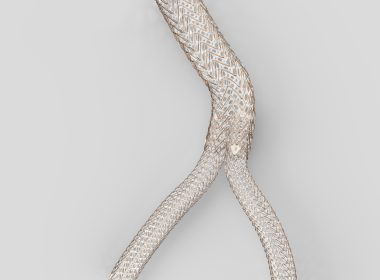
GORE GAINS FDA APPROVAL FOR FIRST DEEP VENOUS STENT INDICATED FOR THE IVC AND ILIOFEMORAL VEINS
The GORE® VIABAHN® FORTEGRA Venous Stent is engineered to offer an optimal balance of conformability and compression resistance for treating a broad range of patients. FLAGSTAFF, Ariz., Jan. 6, 2026 /PRNewswire/ — W. L. Gore & Associates’ medical business (Gore) has announced the FDA…

Spectrumedics Announces CE Mark Approval For Its Coronary Intravascular Lithotripsy System
Sonico-CX provides a safe and effective option for treating calcified coronary lesions. SINGAPORE, Jan. 6, 2026 /PRNewswire/ — Spectrumedics Medical (hereinafter “Spectrumedics”) is pleased to announce that its Sonico-CX Intravascular Lithotripsy (IVL) System has obtained CE Mark…

Shape Memory Medical Secures EU MDR Certification for IMPEDE® Embolization Plug Product Family, Accelerating Global Innovation in Vascular Care
SAN JOSE, Calif.–(BUSINESS WIRE)–Shape Memory Medical Inc., innovator of the proprietary shape memory polymer technology for endovascular applications, announced that its IMPEDE® Embolization Plug product family has received certification as a Class III device under the European Union (EU) Medical Device Regulation (MDR) 2017/745. The IMPEDE Embolization Plug family, previously […]

Pulse Biosciences Announces FDA IDE Approval to Initiate its nPulse Cardiac Catheter Ablation System Study for the Treatment of Atrial Fibrillation
HAYWARD, Calif.–(BUSINESS WIRE)–Pulse Biosciences, Inc. (Nasdaq: PLSE), a company leveraging its novel nPulse™ technology using its proprietary Nanosecond Pulsed Field Ablation™ (nanosecond PFA or nsPFA™) energy, today announced that the U.S. Food and Drug Administration (FDA) has granted approval for the Company’s Investigational Device Exemption (IDE), allowing Pulse Biosciences to […]

HeartSciences Announces FDA 510(k) Submission for MyoVista® wavECG™ Device
Southlake, TX, Dec. 15, 2025 (GLOBE NEWSWIRE) — HeartSciences Inc. (Nasdaq: HSCS; HSCSW) (“HeartSciences” or the “Company”), a healthcare information technology (“HIT”) company advancing the use of ECG/EKGs through the integration of artificial intelligence (“AI”), today announced it has submitted its MyoVista® wavECG™ device to the U.S. Food and Drug Administration (“FDA”) for 510(k) premarket clearance.
Recent

Omada Adds Cholesterol Care to Its Integrated Platform, Addressing the Silent Driver of Cardiovascular Risk

Microbot Medical®’s LIBERTY® Endovascular Robotic System Continues to Expand Market Visibility; Being Featured at an Industry Leading Innovation Conference

AngioDynamics Expands Indications for the NanoKnife System Across Europe, Strengthening Multi-Organ Tumor Ablation Platform
Popular
- No commented posts yet