
September 16, 2024 07:01 PM Eastern Daylight Time NYON, Switzerland–(BUSINESS WIRE)–Edwards Lifesciences today announced the launch in Europe of the SAPIEN 3 transcatheter pulmonary valve implantation (TPVI) system with Alterra adaptive prestent, expanding minimally invasive treatment options to a broader range of patients with congenital heart conditions. “I am proud […]
Coronary/Structural Heart

Heart Failure Society of American Names Journal of Cardiac Failure (JCF) Co-Editors-In-Chief
WASHINGTON, Sept. 17, 2024 /PRNewswire/ — The Heart Failure Society of America (HFSA) Board of Directors has named Anuradha Lala-Trindade, MD and Robert J. Mentz, MD as Co-Editors-in-Chief of the Journal of Cardiac Failure, the official journal of HFSA and the Japanese Heart Failure…
CVRx announces new publication reinforcing the long-term quality of life benefits of Barostim
New data published in JACC: Heart Failure demonstrate durable benefits in MLWHF and EQ-5D quality of life measures in heart failure patients with reduced ejection fraction

Berlin Heals Strengthens Executive Leadership Team
Berlin Heals appoints proven cardiac rhythm management and heart failure leader John Brumfield as Chief Executive Office BERLIN, Sept. 12, 2024 /PRNewswire/ — Berlin Heals Holding AG and its subsidiaries Berlin Heals GmbH and Berlin Heals Corp. which are developing a completely new and…

The Case for Integrating Novel Device-Based Therapies into Guideline Directed Medical Therapy for the Treatment of Heart Failure
WASHINGTON, Sept. 10, 2024 /PRNewswire/ — Novel device-based therapies may overcome limitations of pharmacologic therapies for some patients living with heart failure (HF), indicating that a synergistic approach between the two therapies is ideal for implementation of guideline directed…

BioCardia Announces Closing of Upsized $7.2 Million Public Offering Priced At-The-Market Under Nasdaq Rules
SUNNYVALE, Calif., Sept. 09, 2024 (GLOBE NEWSWIRE) — BioCardia, Inc. [Nasdaq: BCDA], a global leader in cellular and cell-derived therapeutics for the treatment of cardiovascular and pulmonary diseases, today announced the closing of its upsized public offering with participation from management and directors, institutional investors, and certain existing investors of the Company for the purchase and sale of 2,400,000 shares of common stock (or pre-funded warrants in lieu thereof) and warrants to purchase up to 2,400,000 shares of common stock at a combined offering price of $3.00 per share and accompanying warrant, priced at-the-market under Nasdaq rules. The company received aggregate gross proceeds of $7.2 million, before deducting placement agent fees and other offering expenses. The warrants have an exercise price of $3.00 per share, will be exercisable immediately and will expire five years from the issuance date.
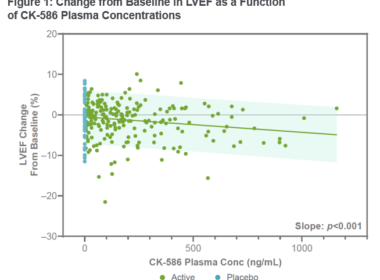
Cytokinetics Announces Data From Phase 1 Study of CK-4021586
Phase 2 Clinical Trial in Patients with Heart Failure with Preserved Ejection Fraction Expected to Begin in Q4 2024

Viz.ai and Cleerly Partner on Heart Disease Evaluation Using Artificial Intelligence
New partnership delivers critical insights to more cardiovascular care teams to identify and define atherosclerosis earlier SAN FRANCISCO – September 4, 2024 — Viz.ai, the leader in AI-powered disease detection and intelligent care coordination, and Cleerly, the company working to create a new standard of care for the diagnosis of heart […]

Renata Medical Announces First Patient to Receive Minima Growth Stent Following FDA Approval
NEWPORT BEACH, Calif.–(BUSINESS WIRE)–Renata Medical, a leader in innovative vascular solutions for young, growing patients, proudly announces a major milestone with the successful implantation of its Minima Growth Stent in the first patient following its recent FDA approval. “Today marks the beginning of something special” Post this “Today marks the […]
ESC Congress 2024: INFINITY-SWEDEHEART Trial of Elixir Medical’s DynamX Bioadaptor Meets Primary Endpoint and Clinical Results Confirm Unique Mechanism of Action
—Largest randomized data set to date on bioadaptor, with a complex patient population of 2,400 compared to a DES— —Primary endpoint of target lesion failure (TLF) non-inferiority at twelve months was met with a low event rate for DynamX despite increased patient complexity— —Pre-specified landmark analysis shows statistically significant reduction […]